
Një sëmundje e trashëguar që mund të deformojë kockat dhe të kufizojë jetën nëse nuk trajtohet me kohë
Sëmundja e rrallë, hipofosfatemia e lidhur me kromozomin X (XLH), është një çrregullim gjenetik që burrat nuk mund ta transmetojnë te djemtë e tyre, por e transmetojnë te të gjitha vajzat. Ndërsa gratë e sëmura e transmetojnë këtë çrregullim në 50% të fëmijëve të tyre, qoftë djem apo vajza, prandaj shpeshherë sëmundja prek më shumë anëtarë të një familjeje. Ky informacion mbështetet nga studimet gjenetike të Institutit Kombëtar të Shëndetit.
Sipas National Organization for Rare Disorders – NORD, kjo sëmundje karakterizohet nga një mutacion në gjenin PHEX në kromozomin X, i cili kontrollon sintezën e një enzime (endopeptidazë) që rregullon nivelin e fosfatit në trup. Si pasojë e këtij mutacioni, fosfati humbet tepricë përmes veshkave dhe traktit tretës, duke shkaktuar uljen e fosfatit në gjak. Kjo gjendje pengon mineralizimin normal të kockave dhe dhëmbëve, duke i bërë ato më të dobëta dhe më të prirura ndaj deformimeve dhe problemeve të tjera.
Humbja e fosfatit si mineral i rëndësishëm bën që kockat dhe dhëmbët të humbasin fortësinë dhe fleksibilitetin e duhur. Kjo manifestohet si përkulje e kockave, sidomos në këmbë, që zakonisht vërehet kur fëmija fillon të ecë. Deformitetet e kockave përparojnë gjatë fëmijërisë dhe mund të bëhen shumë të dukshme gjatë jetës, raporton Mayo Clinic, përcjell Telegrafi.
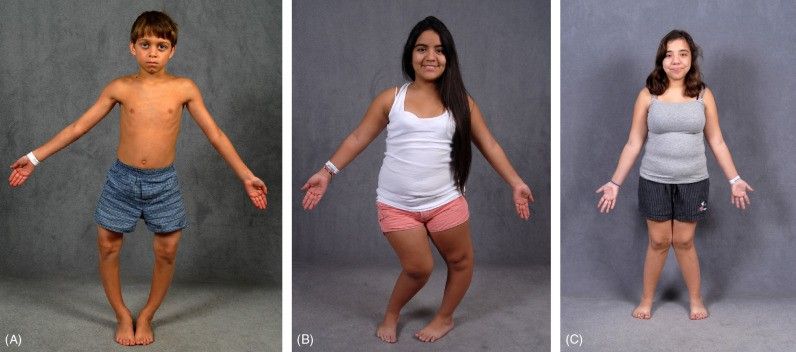
Simptomat kryesore janë ecja e vonuar, vështirësi në vrapim, ecje e paqëndrueshme, dhimbje në kocka dhe nyje, si dhe vështirësi në ngjitjen dhe zbritjen e shkallëve. Gjithashtu, mund të ketë probleme me uljen dhe ngritjen, si dhe vonesë në daljen e dhëmbëve, karies të theksuar për shkak të cilësisë së dobët të smaltit, dhe humbje të hershme të dhëmbëve.
Shenjat e kësaj sëmundjeje ngjajnë me rahitin, por fëmijët nuk reagojnë ndaj terapisë me vitaminë D, që e bën diagnostikimin më të vështirë në mungesë të testeve laboratorike dhe gjenetike. Prandaj, nëse fëmija tregon simptoma të tilla, pavarësisht trajtimit parandalues me vitaminë D, është e rëndësishme të kërkohet vlerësim i specializuar dhe të bëhen analiza laboratorike dhe gjenetike për mutacionin në gjenin PHEX.
Nëse sëmundja nuk trajtohet, mund të shkaktojë komplikime serioze si rritje të ulët disproporcionale, deformime të këmbëve, shtyllës kurrizore dhe duarve, dhimbje kronike, kufizim të lëvizshmërisë së nyjeve dhe rrezik të lartë për fraktura.
Trajtimi përfshin suplementimin me fosfat dhe vitamina D aktive (kalcitriol), që ndihmon në rikthimin e mineralizimit normal dhe parandalimin e komplikimeve. Monitorimi i rregullt laboratorik është i domosdoshëm për përshtatjen e terapisë dhe për të parandaluar efektet anësore. Trajtimi i hershëm ka rëndësi thelbësore për përmirësimin e cilësisë së jetës së pacientit dhe reduktimin e simptomave). /Telegrafi/
